(一)项目基本情况
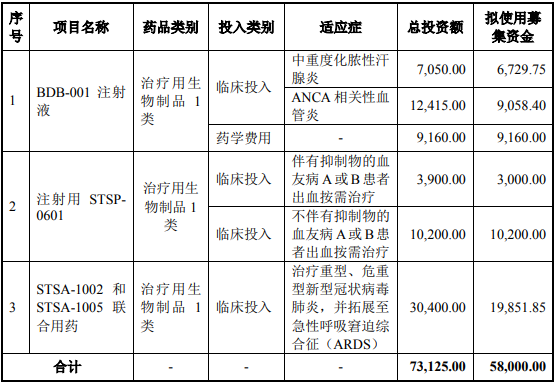
本次拟使用募集资金用于创新药物的临床研究,项目总投资金额为73,125.00万元,拟使用募集资金 58,000.00万元,以满足部分在研产品从现阶段
至 2025 年的研发资金需求。具体如下:单位:万元
各研发项目的具体开展情况如下:
(1)适应症:中重度化脓性汗腺炎(HS)
1)研发背景
化 脓 性 汗 腺 炎 ( HidradenitisSuppurativa , HS ) 又 称 反 常 性 痤 疮(Acneinversanversa,AI),是一种具有家族倾向、反复发作的慢性炎症性皮肤病。其常见临床表现为黑色粉刺、深在性、炎症性皮损,继而形成、疼痛的囊肿、脓肿、窦道、瘢痕等,可伴随或继发多种系统性疾病,如糖尿病、自身炎症性疾病等,多发于腋下、乳房下皱襞、腹部皱襞、腹股沟、臀部、大腿内侧、会阴和肛周等顶泌汗腺分布区域,严重影响患者的生活质量。全球报告 HS不同时期,不同区域 HS 患病率不同。
90 年代 JemecGB 等研究显示全球患病率范围在 1%~4%之间。近代 Zouboulis 等研究显示欧洲平均患病率为 1%。而Cosmatos 等研究发现美国患病率为 0.053%,估计有 146,000~162,000 例患者。法国最近的一项大型流行病学调查报告显示,患病率为 0.97%。英国患病人数估计 10 万左右,每年住院治疗患者超过 2000 人,根据临床登记 HS 患者数量估计患病率为 0.25%~8%。
目前针对HS的治疗手段有限,且往往存在愈合不佳、经常复发的问题。中国目前尚无获批用于治疗 HS 的生物制剂,国外获批的也仅有阿达木单抗(抗TNF-α单抗)一种(FDA2015年9月批准),但仍有大约50%的中度至重度HS患者对阿达木单抗治疗无响应。中重度HS引起的疼痛、慢性化脓和持续恶臭给患者的生活带来巨大的痛苦和经济负担,临床迫切需要开发治疗HS的有效治疗药物。
HS 的发病机理与炎症、细菌感染、遗传、肥胖、吸烟以及机械压力有关。研究显示,多种炎症因子参与 HS 的发病过程,TNF-α、IL-1β、IL-17、IL-12、IL-23 等细胞因子都明显升高。补体系统中的 C5a 具有强烈的趋化作用,可以诱导中性粒细胞、嗜酸粒细胞和单核细胞向炎症部位的移动。此外,C5a 对免疫应答有明显增强作用,可诱导单核细胞分泌 IL-1、IL-6、IL-8 及 TNF-α等细胞因子。
因此提示阻断 C5a 具有对化脓性汗腺炎强有力的治疗潜力,其可以有效控制导致化脓性汗腺炎关键致病因素的炎症因子水平,从而控制HS相关疾病症状。BDB-001 注射液为重组抗人 C5a 人源化单克隆抗体注射液,可以特异性的阻断过敏毒素(C5a)诱导的生物学活性,如中性粒细胞的活化、细胞内颗粒酶的释放、炎症调节因子水平的上升、氧呼吸风暴的爆发等;同时不影响 C5裂解形成 C5a 及膜攻击复合物(MAC)的形成,因此在治疗 HS 上存在巨大的潜力。
公司在中国境内对 BDB-001 独立开展了针对化脓性汗腺炎等适应症的临床研究。BDB-001 注射液治疗中重度化脓性汗腺炎的适应症于 2018 年 7 月在国内取得临床试验批件,2019年 10月第一例受试者入组。2021年 3月国家药品监督管理局药物临床试验登记与信息公示平台公示了单克隆抗体药物 BDB-001 注射液治疗中重度化脓性汗腺炎的 Ib/II期临床试验的相关信息。
2)实施主体、当前研发阶段及预计进展
BDB-001 注射液临床试验项目原先实施主体为舒泰神母公司及全资子公司德丰瑞。截至本预案公告日,舒泰神已与全资子公司德丰瑞签署了吸收合并协议,后续项目实施主体为舒泰神母公司。
BDB-001 注射液项目(适应症:HS)当前研发阶段处于临床 Ib/II 期,临床III期试验预计启动时间为 2023 年四季度。
3)未来前景
目前临床上针对化脓性汗腺炎的药物治疗主要包括抗生素、抗炎药、激素制剂、全身用维 A 酸和中药。尽管目前的药物抗感染、抗炎、抗增生等虽然在短期内获得较好的治疗效果,但是复发的概率仍然很高。国外用于治疗HS的生物制剂也仅有阿达木单抗(Adalimumab),其于 2015 年 09 月经 FDA 批准作为中重度化脓性汗腺炎治疗药物。目前正处在临床研究阶段生物制剂有同样靶向TNF-α的英夫利昔单抗(Infliximab)和依那西普(Etanercept),以及 IL-1受体拮抗剂、IL-23p19 抗体。
即使已获批的阿达木单抗,仍有大约 50%的中度至重度 HS 患者对阿达木单抗治疗无响应。中重度 HS 引起的疼痛、慢性化脓和持续恶臭给患者的生活带来巨大的痛苦和经济负担,中国目前尚无治疗HS的有效药物,临床迫切需要开发治疗 HS 的有效治疗药物。
单克隆抗体药物 BDB001 注射液是针对 C5a 靶点的国内首个、最早进入临床研究的创新药物,有望广泛应用于自身免疫性疾病和感染性疾病,为HS的药物治疗提供新的解决路径。由于该药目前获批临床试验的适应症均无同类型药物上市,市场潜能较大。
另外,近年来研究显示,中性粒细胞异常活化、补体系统异常、多种细胞因子(如 TNF-α、IL-1 和 IL-17 等)分泌异常在中性粒细胞异常活化为特征的皮肤病-嗜中性皮病(neutrophilicdermatoses,ND)的发病中起着关键作用。
这类疾病主要有坏疽性脓皮病、Sweet综合征、白塞病、中性粒细胞性汗腺炎、斯蒂尔病、中性粒细胞性荨麻疹、脓疱型银屑病、IgA 天疱疮、皮肤皱褶无菌性脓疱病、粘液性角化病、婴儿肢端脓疱病等。上述疾病中目前仍有大约1/3的患者一线药物治疗无效。
因此,ND的治疗对于皮肤科医师而言仍是一个巨大的挑战。与BDB-001注射液同靶点同细胞系产品IFX-1(vilobelimab)在坏疽性脓皮病(PG)患者中开展的Ⅱa期临床研究结果显示,高剂量组的 7名患者中有 6名(85.7%)溃疡完全闭合,疗效与患者血浆中 C5a 水平降低相关。IFX-1(vilobelimab)已被美国食品和药物管理局(FDA)和欧洲欧洲药品管理局(EMA)授予治疗 PG的孤儿药资格,InflaRx 公司即将开展 IFX-1(vilobelimab)治疗PG的Ⅲ期临床研究。
BDB-001注射液通过阻断C5a的生物学作用,在一系列中性粒细胞异常活化为特征的皮肤病中亦有可能具有良好的治疗作用,目前尚无同类型药物获批用于上述疾病的治疗,若 BDB-001 注射液后续成功拓展相关适应症,市场潜力巨大。
(2)适应症:ANCA 相关性血管炎
1)研发背景
ANCA 相关性血管炎又称抗中性粒细胞胞浆抗体(ANCA)相关性血管炎,是一组以血清中能够检测到 ANCA 为最突出特点的系统性小血管炎。ANCA 相关性血管炎是一种全身性自身免疫性疾病,是由于补体系统(免疫系统的一部分)的过度激活引起。补体系统的过度激活会进一步激活中性粒细胞,引起炎症并最终破坏小血管。ANCA 相关性血管炎临床表现变化多端,可以累积全身多个器官系统,患者常有发热,疲乏,肌肉关节酸痛等全身炎症反应症状。最常受累的器官是肺和肾。
肾脏损害患者多出现蛋白尿、血尿、各种管型、水肿和肾性高血压等,甚至出现肾功能不全,病情急剧恶化导致死亡。肺部病变主要出现咳嗽、咯血、现呼吸困难或哮喘症状,患者常发生双侧中下肺小叶性炎症,甚至出现肺间质纤维化。其他系统的临床表现包括:患者常出现耳鼻喉的临床症状包括鼻塞,流涕,鼻窦炎,分泌性中耳炎等;神经系统受累最常见的是多发性单神经炎;消化系统表现为消化道出血、胰腺炎以及由肠道缺血引起的腹痛,严重者可出现穿孔;心血管事件如心肌梗死,心力衰竭等的发生率较正常人升高,成为 AAV患者重要的死亡原因。
目前,ANCA 相关血管炎的治疗包括非特异性免疫抑制剂(环磷酰胺或利妥昔单抗),以及联合每日糖皮质激素长期给药,但可能导致显著的临床风险,包括因感染导致的死亡,亟需更好的疗法。与 BDB-001 作用靶点相近的药物Tavneos(Avacopan),作为首个 ANCA 相关血管炎新药,已在日本、美国、欧盟等地陆续上市,其与标准疗法联用,辅助治疗严重活动性抗中性粒细胞胞浆自身抗体(ANCA)相关血管炎,主要包括显微镜下多血管炎(MPA)和肉芽肿伴多血管炎(GPA)。公司 BDB-001 注射液用于 ANCA 相关性血管炎的国内临床试验正在推进中。
2)实施主体、当前研发阶段及预计进展
BDB-001 注射液临床试验项目原先实施主体为舒泰神母公司及全资子公司德丰瑞。截至本预案公告日,舒泰神已与全资子公司德丰瑞签署了吸收合并协议,后续项目实施主体为舒泰神母公司。
BDB-001 注射液(适应症:ANCA 相关性血管炎)当前研发阶段处于临床Ib/II期,预计 2024 年下半年与 CDE沟通并启动临床 III期。
3)未来前景
BDB-001 作为国内首个以 C5a 为靶点治疗 ANCA 相关性血管炎的药物,其作用机制已得到同类药物的验证。如果获批上市,即可有效缓解 ANCA 相关性血管炎患者的症状和体征,减少糖皮质激素的用量,甚至取代糖皮质激素的长
期使用,从而减少其副作用的发生,提高患者生活质量,降低医疗花费,具有巨大的经济效益和社会效益。
(3)BDB-001 注射液项目药学费用
药学费用投入旨在完成 BDB-001 注射液上市前全部所需的药学研究,主要工作内容包含可比性研究、工艺验证、动态核查、现场检查、质量研究、稳定性检验、技术研究、委托检测、注册等环节。
2、注射用 STSP-0601
(1)研发背景
血友病是一种 X 染色体连锁的隐性遗传性出血性疾病,其遗传特点是男性发病,女性携带。每 10 万男孩中有 15 到 20 人发病,此发病率在所调查的不同的种族和地域之间没有差异。发病率以血友病 A 最多,占 85%,余下主要为血友病 B,其他类型血友病较少见。
2018 年 5 月 11 日,国家卫生健康委员会等 5部门联合制定了《第一批罕见病目录》,血友病被收录其中。血友病 A表现为凝血因子 VIII(FVIII)缺乏,血友病 B 表现为凝血因子 IX(FIX)缺乏,均由相应的凝血因子基因突变引起。血友病的临床特征性表现为出血倾向,主要表现为关节、肌肉和深部组织出血等。若反复出血,不及时治疗可导致关节畸形和(或)假肿瘤形成,严重者可危及生命。血友病目前仍无彻底治愈的疗法,凝血因子替代治疗是首选的治疗方法。
血友病 A 患者接受 FVIII 替代治疗后产生的同种中和性抗体成为抑制物。持续合并存在伴抑制物是血友病的严重并发症,将导致血友病患者出血症状更加难控制,致命性出血风险增高。公司研发的凝血因子 X 激活剂“注射用STSP-0601”是国家 I 类治疗用生物制品,凝血因子 X 激活剂“注射用 STSP-0601”是由圆斑蝰蛇毒液分离纯化而得的凝血因子 X 激活剂。该蛋白含有高度复杂的糖基化修饰,所携带糖链多为富含唾液酸的多天线糖型。从作用机理看,
凝血因子 X 激活剂注射用 STSP0601 可特异性地激活凝血因子 X(FX),使活性部位充分暴露生成凝血因子 X激活剂(Fxa),FXa进而与损伤部位激活的血小板、FVa 以及钙离子形成凝血酶原复合物,从而增加凝血酶生成,凝血酶激活损伤部位的血小板和凝血因子 V(FV)、凝血因子(VIII)并通过纤维蛋白原向纤维蛋白的转换形成止血栓,达到帮助出血患者止血的目的。注射用STSP-0601 治疗伴有抑制物的血友病出血机制明确,已有的安全性和有效性数据支持开展注射用 STSP-0601 用于伴有抑制物的血友病 A 或 B 患者出血按需治疗的临床试验。
2019年 4月 30日,公司向国家药品监督管理局提交关于凝血因子 X激活剂“注射用 STSP-0601”的新药临床试验申请,并于 2019年 5月 14日收到国家药品监督管理局《受理通知书》,属于“特殊审批程序”品种;2019 年 7月 31 日,公司收到国家药品监督管理局签发的凝血因子 X 激活剂“注射用 STSP-0601”《临床试验通知书》(CXSL1900045),同意按照提交的方案开展临床试验;2019 年 12 月,关于凝血因子 X 激活剂“注射用 STSP-0601”的药物 I 期临床试验信息于国家药品监督管理局药物临床试验登记与信息公示平台公示(登记号:CTR20191930);
2020 年 1 月第一例受试者入组,已于 2021 年 8 月完成伴抑制物患者的 I 期临床试验,安全性和耐受性良好;2021 年 9 月,公司启动了注射用 STSP-0601 Ib/II期临床试验;2022年 9月 6 日,STSP-0601被 CDE 纳入突破性治疗品种,说明监管机构认可本品已有的临床数据显示出明确的临床优势;CDE 对纳入突破性治疗药物审评程序的品种会采取一系列支持政策,加强指导并促进药物研发进程,优先处理相关沟通交流,加速后续审批流程。
(2)实施主体、当前研发阶段及预计进展
注射用 STSP-0601 项目的实施主体为舒泰神母公司及全资子公司诺维康。“伴有抑制物的血友病 A 或 B 患者出血按需治疗”适应症当前研发阶段处于临床 Ib/II期,预计 2022 年底或 2023年初结束当前阶段;临床 III 期试验预计于 2023 年开展。
“不伴有抑制物的血友病 A 或 B 患者出血按需治疗”适应症当前已收到国家 药 品 监 督 管 理 局 的 《 药 物 临 床 试 验 批 准 通 知 书 》( 通 知 书 编 号 :2022LP01612),该适应症系在伴抑制物临床试验基础上进行的拓展,预计在2023年上半年可以开展有效性临床试验,2023 年下半年可进入临床 III期。
(3)未来前景
按需治疗需要充分考虑患者对于已有治疗手段的可获得性,以及患者自身的经济承受能力。根据《凝血因子 VIII/IX 抑制物诊断与治疗中国指南》,目前用于伴抑制物的血友病患者出血按需治疗的药物有活化凝血酶原复合物(aPCC)和重组人凝血因子 VIIa(rhVIIa)两种。其中,由于我国尚无 aPCC 制剂供应,如果无法获得 rhVIIa,一般使用国产凝血酶原复合物 PCC 止血,PCC 有效止血率仅为 50%,且存在免疫记忆反应、病毒感染和血栓形成等风险;目前国内可用的重组人凝血因子VIIa仅有诺和诺德的进口产品诺其,诺其有效止血率约31-63%,但诺其在国内单支售价约 5000 元,单次出血事件需给药 1~10 次不等,治疗费用约为 3万~30万,限制了临床广泛应用。
因此,研发安全有效且价格可接受的治疗药物是国内伴有抑制物的血友病患者急性出血发作亟待解决的临床需求。此外,随着注射用 STSP-0601 项目研发的推进,将瞄准满足不伴有抑制物的血友病患者的临床需求,进一步拓宽市场空间。
本项目是具有自主知识产权的国家I类创新生物药物,该产品结构复杂,生产工艺和质量控制技术难度高,具有较高的研发技术门槛。其研发的成功将为血友病患者提供安全、有效且经济可接受的治疗药物,从而大幅提高血友病患者接受治疗的比例,减轻患者负担,创造社会效益。
3、STSA-1002 和 STSA-1005 联合用药
(1)研发背景
急性呼吸窘迫综合征(ARDS)是一种在短时间内 (1 周内)发生的急性、弥漫性的炎症性肺损伤,由严重感染、创伤、休克等各种肺内外致病因素导致,临床表现为呼吸窘迫、顽固性低氧血症和呼吸衰竭,为常见的危及人类健康的呼吸系统重症表现之一。
多种危险因素可诱发 ARDS,主要包括:1)直接肺损伤因素:严重肺部感染,胃内容物吸入,肺挫伤,吸入有毒气体,淹溺、氧中毒等;2)间接肺损伤因素:严重感染,严重的非胸部创伤,重症急性胰腺炎,大量转输血,体外循环,弥散性血管内凝血等。SARS-CoV、MERS-CoV、SARS-CoV-2 以及流感病毒引发的 ARDS 也是导致患者死亡的主要原因之一。
ARDS 早期的特征性表现为肺毛细血管内皮细胞与肺泡上皮细胞屏障的通透性增高,肺泡与肺间质内积聚大量的水肿液,其中富含蛋白及以中性粒细胞为主的多种炎症细胞。中性粒细胞黏附在受损的血管内皮细胞表面,进一步向肺间质和肺泡腔移行,释放大量促炎介质,如炎症细胞因子等,参与中性粒细胞介导的肺损伤。除炎症细胞外,肺泡上皮细胞以及成纤维细胞也能产生多种细胞因子,从而加剧炎症反应过程。
凝血和纤溶紊乱也参与了 ARDS 的病程,ARDS 早期促凝机制增强,而纤溶过程受到抑制,引起广泛血栓形成和纤维蛋白的大量沉积,导致血管堵塞以及微循环结构受损。流行病学调查显示,ARDS 是临床常见危重症。2005 年研究显示 ARDS 发病率为每年 59/10 万,较以往发病率显著提高,明显增加社会和经济负担,甚至可与胸部肿瘤、获得性免疫缺陷综合征(AIDS)、哮喘或心肌梗死等相提并论。
近年来,SARS-CoV、MERS-CoV、SARS-CoV-2 以及流感病毒世界各地的爆发性传播导致了大量患者死亡,尤其是合并有基础疾病的老年人,因此,针对 ARDS 的研究具有较强的现实意义,ARDS 的研究领域也不局限于 COVID-19,可拓展至更多的肺部感染因素。目前,STSA-1005 和 STSA-1002 单药的非临床研究比较充分,STSA-1005 注射液和 STSA-1002 注射液已开展的单药 I 期临床研究显示安全性良好,STSA-1002 和 STSA-1005 联合用药能够抑制过度激活的髓性细胞(单核/巨噬细胞、中性粒细胞),且 STSA-1005 可减少髓性细胞的骨髓动员,STSA-1002 对改善患者血栓并发症存在益处,具有协同的潜力,为两药联合治疗重型、危重症新型冠状病毒肺炎及后续拟拓展到 ARDS 的临床研究提供了理论依据。
(2)实施主体、当前研发阶段及预计进展
STSA-1002 和 STSA-1005 联合用药的实施主体为舒泰神母公司和全资子公司 STAIDSON BIOPHARMA, INC.,已于 2022 年 8 月取得临床试验批准通知书(适应症:治疗重型、危重症新型冠状病毒肺炎),目前处于临床I期阶段,预计在 2023年上半年完成健康受试者临床 I期试验,之后将向 FDA申请和沟通后续适应症拓展至 ARDS 适应症的具体事宜,II 期预计最快在 2023 年下半年开展,最快 2024 年下半年进入 III期。
(3)未来前景
ARDS 患者仍以机械通气作为主要支持治疗方式,但这一手段通过呼吸机控制气压使肺泡扩张,可能造成肺泡壁损伤,进一步诱发机械通气相关性肺损伤。药物治疗主要包括液体管理、糖皮质激素、肺泡表面活性物质等,但仍存在未满足的临床需求,研发安全、有效治疗 ARDS 药物仍具有重要意义。
(二)项目实施的必要性
1、保障创新药物研发投入,加快创新药物研发落地
公司所处的生物创新药行业属于资金密集型行业。创新药临床试验监管严格,过程周期较长,试验复杂,对公司的资金投入有着较高要求。本次募集资金用于重点在研产品的临床试验推进,保障上述在研产品按照既定的临床试验计划,进入临床治疗应用,获得临床试验结果,完成创新药物的完整研发过程。本项目成功实施后,将有效保障公司创新药物的研发进程,拓展公司在研药物的临床试验广度和深度,为公司实现更多可商业化的产品奠定基础。
2、增强公司核心竞争力,巩固公司竞争优势
公司所处的生物创新药行业属于技术密集型行业,产品生命周期有限,技术迭代升级较快。因此,研发是创新药企业的发展基石和核心竞争力。创新药企业为保持竞争优势,不断储备拓展研发管线产品,增强研发的深度和广度,为持续增长、增强核心竞争力提供保障。
公司自设立以来,持续进行创新生物药物产品的研发,在发现及开发创新大分子生物药的领域能力突出,能够独立进行发现及开发生物药所需的靶点评估、机制研究、验证、临床在研药品筛选以及功能学验证等全流程发现及开发生物药。本次募投项目对应的在研产品在作用机理和研发工艺方面均处于国内领先水平,国内同靶点产品稀缺,公司一旦研发成功将有效提升产品体系的竞争力,创造新的增长点,并巩固公司的技术优势,从而为公司的可持续发展提供创新动力。
(三)项目实施的可行性
1、符合国家产业政策和行业发展方向
中国从多方面颁布鼓励政策,支持并鼓励生物药的研发,如 2017 年 10 月颁布了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,通过改革临床试验管理和加快审评审批等多方面鼓励生物药创新和生物类似药研发,2020 年,我国就创新药的优先审评审批、专利补偿、数据保护、临床试验规范(GCP)等颁布或者修订了一系列的政策或规则,以支持创新药的研发及生产。
经修订的《药品注册管理办法》《药品生产监督管理办法》《药物临床试验质量管理规范》及《生物制品注册分类及申报资料》陆续生效,对新药研发、注册流程、临床试验管理以及生产管理等环节进行了改革。2020 年 7 月,国家药品监督管理局发布《突破性治疗药物审评工作程序(试行)》,对纳入突破性治疗药物程序的药物,CDE 将优先配置资源进行沟通交流,加强指导并促进药物研发。2021 年 5 月,《关于全面加强药品监管能力建设的实施意见》发布,要求提高完善创新药的审评机制。2021 年 6 月,新修订的《中华人民共和国专利法》生效,其中对在中国获得上市许可的新药相关发明专利给予了专利权期限补偿,这一系列政策均对国内创新药的研发及上市生产销售带来了促进作用。
2022 年 1 月 30 日,《“十四五”医药工业发展规划》正式出台,把坚持创新引领作为基本原则,把创新作为推动医药工业高质量发展的核心任务,加快实施创新驱动发展战略,构建开放创新生态,提高创新质量和效率,加快创新成果产业化,为医药工业持续健康发展打造新引擎。
2、技术实施具有可行性
公司是一家具有创新能力,涵盖新药研发、生产和销售的全产业链创新性生物制药企业,是国家级高新技术企业。
公司经过多年积累,创新药物研发体系构建基本完成,具有丰富的技术储备、专业的研发技术团队,对药物设计、筛选、细胞系构建、临床前评价、临床评价、药品生产和质量控制、产业化等进行系统化研究开发和迭代升级。蛋白药物方向,建立了多种达到业界领先水准的候选药物筛选体系、计算机辅助药物结构优化平台、蛋白药物工艺开发和中试放大平台、质量研究及控制平台;基因治疗药物/细胞治疗药物方向,建立了递送载体筛选及评价体系、工艺开发
中试放大平台、质量研究及控制平台。同时,公司根据疾病治疗领域设置对应的药理毒理研究团队和临床研究及运营团队。以上各技术平台的建立,有效支撑并推动公司的创新药物研究工作。在自身研发的基础上,公司积极开展与第三方专业机构的技术合作,通过整合内外部研发资源,完成新产品开发和技术成果的转化。
3、人才可行性
公司产品和技术的持续性创新是以人为基础的,组建了一支多元化的、具有国际视野并具备扎实的专业素养和丰富的药物开发经验的专业团队。公司研发人员多毕业于国内外知名医学、药学、生物学专业院校,或具有国际化大型制药企业工作经验,截至 2022年 9月 30日,公司现有研发人员 263人,占员工总人数的比例达到 32.92%。其中硕士以上学历人员 166 人,占研发人员总人数的比例为63.12%;博士以上学历人员45人,占研发人员总人数的比例为17.11%。公司对研发团队的高度重视及对研发投入逐年增长,为研发项目和技术平台的升级提供了持续的人才保障和资金保障。
4、管理可行性
公司具备完善的生产和质量管理体系、供应商管理体系、人力资源管理体系,拥有一支稳定、凝聚力强,并拥有丰富的管理经验和多年生物制药的行业经验的团队。为了提高创新能力,加强新技术、新产品、新工艺的研究开发和管理,加快技术积累和产品升级,公司制定了完善的研发体系,在项目立项管理、药品注册管理、鼓励技术创新、技术成果保护等方面建立了明确的规章制度。此外,公司与国内外知名的医药企业、医疗机构建立了稳定的合作关系和成熟的合作机制,通过合作协议明确约定了研发过程中各方的合作方式、工作职责、保密义务、款项支付、成果分配,在临床治疗方面持续开展合作。
(四)项目与现有业务或战略的关系
公司是一家具有创新能力,涵盖新药研发、生产和销售的全产业链创新性生物制药企业。本次募集资金投资项目紧紧围绕公司主营业务展开,是对公司现有研发能力的提升和扩充,为公司实现中长期战略发展目标奠定坚实的基础。
随着本次募投项目的实施,将进一步推进公司创新药物的研发进程、丰富在研药物产品管线、增强研发实力,拓展公司在研药物的临床试验广度和深度,进一步提升公司的核心竞争力,为公司实现更多可商业化的产品奠定基础。
(五)项目效益分析
本次募投项目的实施,将加快公司在研新药研发进程,推动在研产品的尽快上市。由于药品需要完成临床试验、获得新药注册批件和生产文号后再进行商业化,还涉及产品生产、销售推广等多个领域,因此本次募投项目无法单独直接计算经济效益。
(六)资金缺口的解决方式
本项目计划投资规模为 73,125.00万元,其中计划募集资金总额为 58,000.00万元。若本次发行募集资金不能满足公司项目的资金需要,公司将利用自筹资金或通过其他融资方式解决不足部分。
(七)项目涉及审批、批准或备案事项的情况
根据国家发改委发布的《企业投资项目核准和备案管理办法》,本次募集资金拟投入的创新药物研发项目主要为处于临床研究阶段实施的药物研发工作,不属于需要发改委备案的固定资产投资项目;同时,上述创新药物研发项目不涉及生产建设活动,不属于根据《中华人民共和国环境影响评价法》和《建设项目环境影响评价分类管理名录》等相关法律法规的规定需要进行环境影响评价的建设项目。
此报告为正式报告摘取部分。需编制政府立项、银行贷款、投资决策等用途可行性研究报告咨询思瀚产业研究院。








 无锡国家数字电影产业园三期项目可行性研究报告
无锡国家数字电影产业园三期项目可行性研究报告 华侨城文旅装备产业园工程建设投资项目可行性研究报告
华侨城文旅装备产业园工程建设投资项目可行性研究报告 美国亚利桑那州-记忆绵床垫生产基地扩建项目可行性研究报告
美国亚利桑那州-记忆绵床垫生产基地扩建项目可行性研究报告 江西宜春-高能量密度动力储能(方形)锂电池研发产业化项目可行性研究报告
江西宜春-高能量密度动力储能(方形)锂电池研发产业化项目可行性研究报告 水晶光电-台州智能终端用光学组件技改项目可行性研究报告
水晶光电-台州智能终端用光学组件技改项目可行性研究报告 广西钦州-中伟股份北部湾产业基地三元项目一期可行性研究报告
广西钦州-中伟股份北部湾产业基地三元项目一期可行性研究报告 中国天津-毫米波雷达研发中心建设项目可行性研究报告
中国天津-毫米波雷达研发中心建设项目可行性研究报告 中国重庆-国储珞璜智慧物流园项目可行性研究报告
中国重庆-国储珞璜智慧物流园项目可行性研究报告